In the spring of 2001 I researched and wrote this report on Huntington's Chorea for my Biology class.
Huntington's chorea-also less accurately known as Huntington's disease-is a severe genetic disorder of the nervous system, responsible for changes in the brain that have severe physical and mental effects.
Huntington's chorea was named for George Huntington, an American physician who first described the disease in 1872. A chorea is any number of disorders characterized by brief, rapid, uncoordinated movements. The word was derived from the Greek word for 'dance.' Huntington's chorea is hereditary, which means is can be transferred from parent to child through a specific gene or group of genes. Because the gene is dominant, Huntington's chorea cannot skip generations; if the disease is transmitted from a parent it is certain to develop in the child, and only those who have developed the disease can pass it along. If one of a child's parents has the Huntington's gene, the child usually has a fifty percent chance of receiving the gene, unless by some chance that parent received the gene from both his or her parents, in which case he or she will be homozygous dominant for the disease and certain to pass it along to his or her child.
Huntington's chorea destroys brain cells, upsetting the balance of chemicals in the brain and causing involuntary body movements, mental disturbances, and eventually, death (usually from pneumonia, choking, or other complications. Some patients may become depressed enough to commit suicide).
Huntington's chorea probably affects roughly one out of every ten thousand people in the United States, but this is difficult to say for certain, because Huntington's chorea is often misdiagnosed as schizophrenia or other similar diseases. There is no cure for the yet, only various methods for detection and drugs to ease the symptoms.
In the 1980's, certain genetic markers were discovered for the disease, making detection easier. At this time the only test for Huntington's chorea (to determine whether or not the gene was present in someone who had not yet developed the disease) required tissues from several family members, including one who had the disease and one who did not. In March of 1993, however, scientists discovered the gene, located on the fourth chromosome, which made possible a new predictive test, requiring only a blood sample from the patient. The disease itself is caused by a mutation in this chromosome, which causes extra copies of the gene involved to be produced. This disrupts the normal manufacturing of proteins, which is responsible for the death of nerve cells in the basal ganglia.
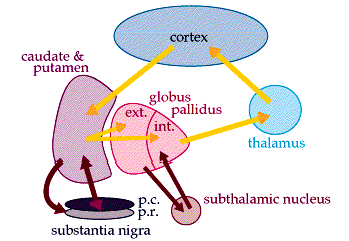
The basal ganglia are a collection of nuclei deep in the white matter of the cerebral cortex (in the frontal lobe) that modifies movement on a minute-to-minute basis. The basal ganglia is divided into several sections, called the caudate, the putamen, the nucleus accumbens, the globus pallidus, the substantia nigra, and the subthalamic nucleus. The caudate and putamen receive input from the motor cortex and send it on to both the substantia nigra and the globus pallidus. (See picture below).
Part of the substantia nigra, the substantia nigra pars compacta, sends the information it receives right back to the caudate and putamen, but also produces dopamine, a neural transmitter critical for normal movement. The other part, the substantia nigra pars reticulata, sends the information outside the basal ganglia to control head and eye movements.
The globus pallidus receives the input from the caudate and putamen and sends it on to-and receives it back from-the subthalamic nucleus. A portion of the globus pallidus, the globus pallidus interna, then sends the motion-inhibiting commands out of the basal ganglia altogether, into the thalamus, and back to the motor cortex.
The nerve cells that die in the basal ganglia are located in the caudate and putamen. These cells produce two neurotransmitters, the quantities of which are then depleted. Meanwhile, a third neurotransmitter (dopamine) builds up in this area. All these changes disrupt the complex cycle of information within the basal ganglia, so that it can no longer perform its function of inhibiting motion in the body, which causes the involuntary movements of Huntington's chorea.
The degeneration of the caudate and putamen usually appears in mid-life, around the ages of thirty-five to fifty-five, but there have been cases where the first symptoms appear as young as childhood or as late as old age.
The very earliest symptoms of the disease can be slight clumsiness, restlessness, or what appear to be minor problems with coordination. Later on, muscles in the face and hands may begin to move involuntarily, including pouting of the lips, irregular raising of the eyelids, and puffing out of the cheeks. Speech may be slurred. As the disease progresses, these involuntary movements spread throughout the whole body. Head, shoulders, arms, and legs jerk uncontrollably. The uncontrolled movements combine with voluntary movements to produce a lurching, dance like appearance when walking.
Meanwhile, many mental changes may be taking place. The earliest symptoms can include dullness, irritability, apathy, and carelessness, especially about personal grooming. Judgment is impaired. The disease eventually progresses to memory loss, dementia, and manic depression. Patients may finally lose all muscle control and mental ability.
Huntington's chorea can progress from ten to twenty years before death finally occurs.
"Fact Sheet: Huntington's Disease." Family Caregiver Alliance. Online. Internet. Available WWW: http://www.caregiver.org/factsheets/diagnoses/huntingtons.html
"Basal Ganglia and Cerebellum." Washington University Department of Anatomy and Neurobiology. Online. Internet. Available WWW: http://thalamus.wustl.edu/course/cerebell.html
"Chorea." Britannica.com. Online. Internet. Available WWW: http://www.britannica.com/eb/article?eu=84528&tocid=0
"Huntington's Chorea." Grolier International Encyclopedia. Danbury: Grolier Incorporated, 1993. 10:315.
"Huntington's Chorea." The World Book Multimedia Encyclopedia, Version 3.00, IBM/World Book, 1999.